
RNA-seq Bioinformatic Workflow
Pocillopora spp. RNAseq QC, Alignment, Assembly Bioinformatic Pipeline
Goal
The following document contains the bioinformatic pipeline used for cleaning, aligning and assembling our raw RNA sequences. These commands were compiled into bash scripts to run on the bluewaves server and are available on the Molecular Underpinnings of Enhanced Thermal Performance due to Chronic Low Nutrient Enrichment Repository
Author: Hollie Putnam and Danielle Becker Last Updated: 2021/04/14 Data uploaded and analyzed on the URI Putnam Lab bluewaves server.
Project overview
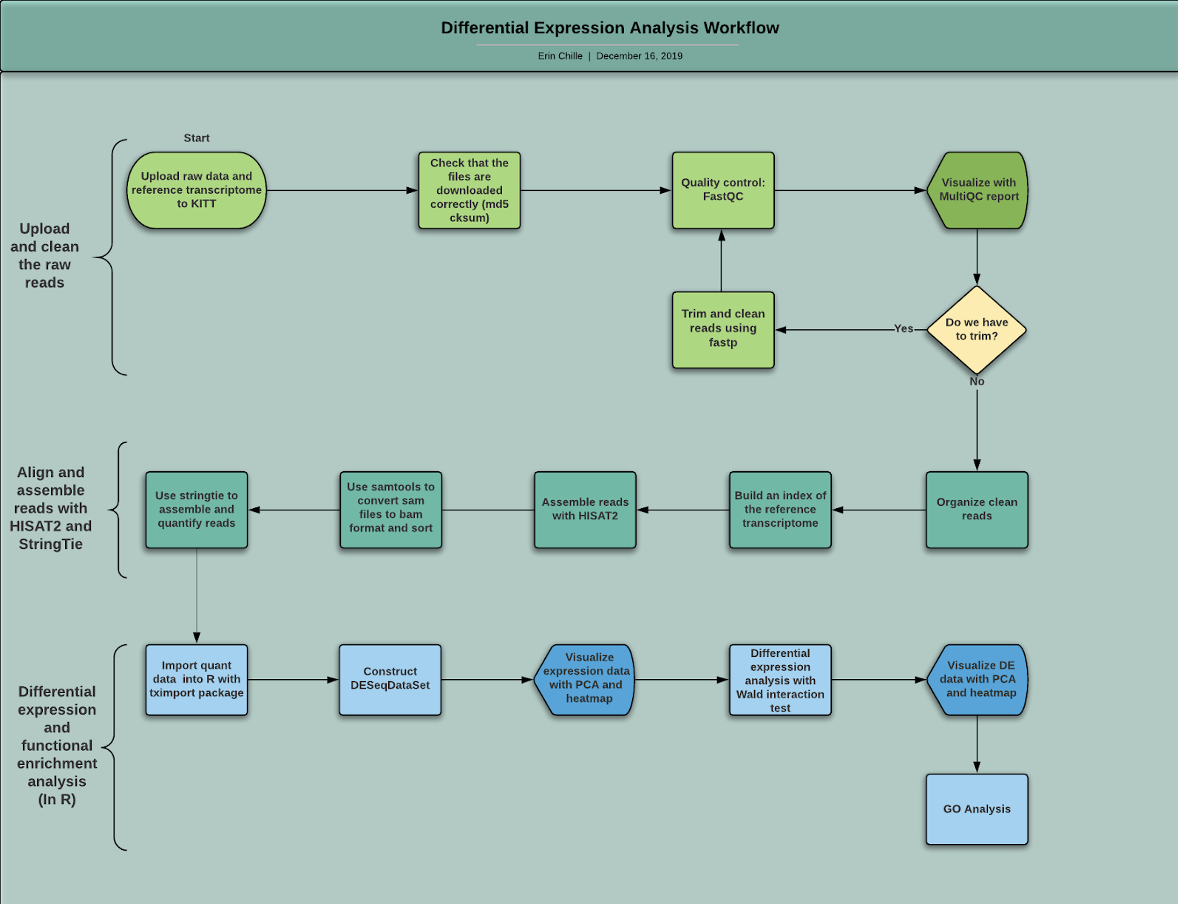
Bioinformatic tools used in analysis: Quality check: FastQC, MultiQC Quality trimming: Fastp Alignment to reference genome: HISAT2 Preparation of alignment for assembly: SAMtools Transcript assembly and quantification: StringTie
Check for required software on bluewaves
Update software to latest version
- fastqc
- MultiQC
- fastp
- HiSat2
- Samtools
- StringTie
- gffcompare
- Python
Prepare work space
- Upload raw reads and reference genome to server
- Assess that your files have all uploaded correctly
- Prepare your working directory
- Install all necessary programs
Upload raw reads and reference genome to server
This is done with the scp or “secure copy” linux command. SCP allows the secure transferring of files between a local host and a remote host or between two remote hosts using ssh authorization.
++Secure Copy (scp) Options++:
- -P - Identifies the port number
- -r - Recursively copy entire directories
scp -r -P xxxx <path_to_raw_reads> danielle_becker@bluewaves.uri.edu:<path_to_storage> scp -P xxxx <path_to_reference> danielle_becker@bluewaves.uri.edu:<path_to_storage>
1) Obtain Reference Genome
cd /data/putnamlab/REFS/
mkdir Pverr
Genome scaffolds
wget http://pver.reefgenomics.org/download/Pver_genome_assembly_v1.0.fasta.gz
Gene Models (CDS)
wget
http://pver.reefgenomics.org/download/Pver_genes_names_v1.0.fna.gz
Gene Models (Proteins)
wget
http://pver.reefgenomics.org/download/Pver_proteins_names_v1.0.faa.gz
Gene Models (GFF)
wget
http://pver.reefgenomics.org/download/Pver_genome_assembly_v1.0.gff3.gz
Check to ensure data transfer of genome files
Core files MD5 hash
Genome scaffolds fb4d03ba2a9016fabb284d10e513f873
Gene models (CDS) 019359071e3ab319cd10e8f08715fc71
Gene models (proteins) 438f1d59b060144961d6a499de016f55
Gene models (GFF3) 614efffa87f6e8098b78490a5804c857
Miscellaneous files MD5 hash
Full transcripts 76b5d8d405798d5ca7f6f8cc4b740eb2
On Bluewaves
Pver_genes_names_v1.0.fna.gz 019359071e3ab319cd10e8f08715fc71
Functional annotation
Functional Annotation File (xlsx) Link
Make folder structure
mkdir data
cd data
ln -s ../../../../../KITT/hputnam/20201209_Becker_RNASeq_combo/combo/*.fastq.gz ./raw/
ln -s ../../../../../REFS/Pverr/ ./refs/
Download files
Path where we stored the RAW fastq.gz files
# 2) Check file integrity
a) Check to make sure you have all of your files, and that they all follow the same naming convention. There should be 64 fastq.gz files. First we will look at our list of files in our read storage directory, and then we will count the number of fastq.gz files.
ls -1 | wc -l
#### Verify data transfer integrity with md5sum
b) Check to make sure the files downloaded correctly using the md5sum command. First store the md5checksum in a file then verify the contents of the new md5sum file.
nano /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/scripts/check_transfer.sh
#!/bin/bash ###creating slurm script #SBATCH -t 24:00:00 ###give script 24 hours to run #SBATCH –nodes=1 –ntasks-per-node=1 ###on server, different nodes you can use for processing power, node just do one task #SBATCH –export=NONE #SBATCH –mem=100GB ###in server allocate 100GB amount of memory #SBATCH –account=putnamlab ###primary account #SBATCH -D /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/raw ###path
md5sum /dbecks/Becker_E5/Becker_RNASeq/data/*.gz > URIcheckmd5.md5
md5sum /data/putnamlab/KITT/hputnam/20201209_Becker_RNASeq_combo/combo/*.gz > URIcheckmd5.md5
sbatch check_transfer.sh
###Submitted batch job 1816196 20201230
- [x] ++Md5 Output++:
All files "OK"
### Checksum from Genewiz
317dd03e9704a73347c5ccabb86d1e18 C17_R1_001.fastq.gz b6368b2789cfacc8e8adbadf45d6c533 C17_R2_001.fastq.gz adb1af7c62b309f7100117f50ebe846d C18_R1_001.fastq.gz 776842109ee56cd4619d5dec08fe005e C18_R2_001.fastq.gz a6d48537ec697d2040798a302bfa0aa1 C19_R1_001.fastq.gz 9b46b63800d83940b53196dccd4eda53 C19_R2_001.fastq.gz e2a63dca87aa53b4756d51edb02b377f C20_R1_001.fastq.gz b2752d8b60c97f754cc24ededc1bf053 C20_R2_001.fastq.gz f659265a858c029bfb4d2764da76701a C21_R1_001.fastq.gz 712ffc2eb87aadd42d50f9c10c7c9b81 C21_R2_001.fastq.gz 3fe2c3f4c39c06adca53440c037d48fe C22_R1_001.fastq.gz ff775c4de3ad8a585f64367cc78e467d C22_R2_001.fastq.gz 2f4f97aa4dd101b5a14ab0a4284f0816 C23_R1_001.fastq.gz aabac96314290241fda7e7413d716b2c C23_R2_001.fastq.gz 94fa7191998ac59d2e89851b5fb15431 C24_R1_001.fastq.gz 921198dae25a3c4515be0d534281a685 C24_R2_001.fastq.gz feb863a003dfff84ca3868ec5b674e02 C25_R1_001.fastq.gz 3ae4179cd04cdf04bd5873ba5d6a0534 C25_R2_001.fastq.gz 225bafe67a42557b7a34ac201cf88273 C26_R1_001.fastq.gz 15aee2823967a782a859c039610b0b23 C26_R2_001.fastq.gz 82ec398d754d52fef140a325c80ee289 C27_R1_001.fastq.gz 8aebf98f2af591cf8a1149f263f6d86d C27_R2_001.fastq.gz 1bf51f4961df4b7da8f284cd7202f6d3 C28_R1_001.fastq.gz 1b4cc1e1be421387abfd7da8d490235b C28_R2_001.fastq.gz f02e752d98e9a0168286becf52203c0f C29_R1_001.fastq.gz fd280e79ecaa4a51a1670293c742d5fd C29_R2_001.fastq.gz 27153ad98aa8735c3474bd72dfe041cd C30_R1_001.fastq.gz 8d977f9ebebea4c26c0cfc9bf7033a9b C30_R2_001.fastq.gz b6a2cf9d02329c3c73c4ec02c1441660 C31_R1_001.fastq.gz 52cdaf65c3cb976c168478678131066f C31_R2_001.fastq.gz cdfc0509137d2b8624a236b2ded04a47 C32_R1_001.fastq.gz 9148b0e6bbc9b7520c7e1c6caae13fc8 C32_R2_001.fastq.gz 8854b4f07a80e5c0e49a136bb5c76e75 E10_R1_001.fastq.gz 1cccd8da94d4a8a79f1681edc9349166 E10_R2_001.fastq.gz e7277e7ac17b4643e748e1a3e00dfb04 E11_R1_001.fastq.gz 3ff35b0852ea83593dd438dcbb2deddd E11_R2_001.fastq.gz 50ab9cee762e076b5183de2c8fc66f02 E12_R1_001.fastq.gz e188fc0f1f0ecd1feb581dd01574667c E12_R2_001.fastq.gz af969bcc9b91dfd59c48f8cb87fb6317 E13_R1_001.fastq.gz 7759be7b6477a401e4ffcf385b80f51e E13_R2_001.fastq.gz c0d20187ee3403b1a1e4687afbd2fd91 E14_R1_001.fastq.gz 80f122aca444d7ebdfc153d73666cf4b E14_R2_001.fastq.gz a7632155416f4de7c80f0f1466c21457 E15_R1_001.fastq.gz 9eec50018582ad56cc2ad05970da7b90 E15_R2_001.fastq.gz 1f782733b62f17958a813d09793922f0 E16_R1_001.fastq.gz 91f58b438ae17444c510a6636bf244ce E16_R2_001.fastq.gz bf0038545deef9b4729b8b322ab5f33b E1_R1_001.fastq.gz f3e8fab360a0986397ad1e6dd6a85afd E1_R2_001.fastq.gz 13cc2a96b34654c62eae372ebd8109ea E2_R1_001.fastq.gz a55437f2ba10eac88e70994622c20cc2 E2_R2_001.fastq.gz be0aa42447cfa4e52e47e4c1587f07f6 E3_R1_001.fastq.gz 0dd2317421ce84e9ec1d9a029752a9b3 E3_R2_001.fastq.gz 44b7219ff47447522e24bb6d10ec871c E4_R1_001.fastq.gz 6a9249b714e0a1596bad222d94bcca09 E4_R2_001.fastq.gz beedc3c1f5d7918166f8f26d867cdb12 E5_R1_001.fastq.gz f45e1969d25e3e07b993672b3a6ef8f7 E5_R2_001.fastq.gz 3792a2a487030d0b3c4ad7fe95bff398 E6_R1_001.fastq.gz 004eb857e060728c619262f7287ec273 E6_R2_001.fastq.gz fd8d4df782b0142bea08adf04edbe835 E7_R1_001.fastq.gz c21ffd9c4d24181ff6f5e8bf02c83a95 E7_R2_001.fastq.gz 0852370e047f41a248ca7e7014ad88dc E8_R1_001.fastq.gz b6e5da4446b5bccb63e46661e9b1e293 E8_R2_001.fastq.gz 1ecdc2f728e6ce233c9495a2c3bb97f5 E9_R1_001.fastq.gz be19f114b314fdc30dd39962aa12a3dc E9_R2_001.fastq.gz
c) Cross-reference the checksum document from GENEWIZ with the data we have on our computer
with a small amount of files, able to first cross-check that the sequences matched between both files on the desktop
used the code below in terminal to cross-check the files and compare for sanity check
d) Count number of reads per file using the code after @ in fastq.gz files (e.g.,@GWNJ).
zgrep -c “@GWNJ” *.gz > raw_seq_counts
- [x] ++Raw Read Counts++:
C17_R1_001.fastq.gz:25158606 C17_R2_001.fastq.gz:25158606 C18_R1_001.fastq.gz:22733345 C18_R2_001.fastq.gz:22733345 C19_R1_001.fastq.gz:24846067 C19_R2_001.fastq.gz:24846067 C20_R1_001.fastq.gz:24030431 C20_R2_001.fastq.gz:24030431 C21_R1_001.fastq.gz:16484060 C21_R2_001.fastq.gz:16484060 C22_R1_001.fastq.gz:22990550 C22_R2_001.fastq.gz:22990550 C23_R1_001.fastq.gz:20905338 C23_R2_001.fastq.gz:20905338 C24_R1_001.fastq.gz:22578178 C24_R2_001.fastq.gz:22578178 C25_R1_001.fastq.gz:29417106 C25_R2_001.fastq.gz:29417106 C26_R1_001.fastq.gz:23267238 C26_R2_001.fastq.gz:23267238 C27_R1_001.fastq.gz:24990687 C27_R2_001.fastq.gz:24990687 C28_R1_001.fastq.gz:23396439 C28_R2_001.fastq.gz:23396439 C29_R1_001.fastq.gz:17900262 C29_R2_001.fastq.gz:17900262 C30_R1_001.fastq.gz:26361873 C30_R2_001.fastq.gz:26361873 C31_R1_001.fastq.gz:24642131 C31_R2_001.fastq.gz:24642131 C32_R1_001.fastq.gz:27076800 C32_R2_001.fastq.gz:27076800 E10_R1_001.fastq.gz:29131006 E10_R2_001.fastq.gz:29131006 E11_R1_001.fastq.gz:18568153 E11_R2_001.fastq.gz:18568153 E12_R1_001.fastq.gz:27805087 E12_R2_001.fastq.gz:27805087 E13_R1_001.fastq.gz:24455094 E13_R2_001.fastq.gz:24455094 E14_R1_001.fastq.gz:22630044 E14_R2_001.fastq.gz:22630044 E15_R1_001.fastq.gz:23796710 E15_R2_001.fastq.gz:23796710 E16_R1_001.fastq.gz:29523059 E16_R2_001.fastq.gz:29523059 E1_R1_001.fastq.gz:25368402 E1_R2_001.fastq.gz:25368402 E2_R1_001.fastq.gz:23770610 E2_R2_001.fastq.gz:23770610 E3_R1_001.fastq.gz:25641209 E3_R2_001.fastq.gz:25641209 E4_R1_001.fastq.gz:21751368 E4_R2_001.fastq.gz:21751368 E5_R1_001.fastq.gz:16381619 E5_R2_001.fastq.gz:16381619 E6_R1_001.fastq.gz:24937261 E6_R2_001.fastq.gz:24937261 E7_R1_001.fastq.gz:24020166 E7_R2_001.fastq.gz:24020166 E8_R1_001.fastq.gz:23675842 E8_R2_001.fastq.gz:23675842 E9_R1_001.fastq.gz:25068848 E9_R2_001.fastq.gz:25068848 ======= /data/putnamlab/KITT/hputnam/20201209_Becker_RNASeq_combo/combo/md5sum_list.txt
#### Prepare your working directory
a) Create your working directory. Within your working directory make subdirectories for scripts, data, and output. Enter the data directory and make a subdirectory to place raw reads and reference files.
mkdir Becker_RNASeq cd Becker_RNASeq
mkdir scripts mkdir data
cd data mkdir raw mkdir ref mkdir counts
b) Create symbolic links to raw reads and reference sequences.
ln -s
#### Install all necessary programs
a) Install programs within your conda environment, when possible.
b) Create and activate a conda environment. Must have [miniconda](https://docs.conda.io/en/latest/miniconda.html) installed.
conda create -n Becker_RNASeq conda activate Becker_RNASeq
c) Install all necessary programs within your conda environment
conda install fastqc conda install multiqc conda install fastp conda install hisat2 conda install samtools
d) The version of StringTie available on Bioconda is not the most recent version (v2.1.0). The version installed in conda (v2.0) has errors when running with the '-e' option that we need for this next step in StringTie. We will have to install StringTie outside of the conda environment. The following commands will install the latest version and test the binary. This only took about 3 min to run.
git clone https://github.com/gpertea/stringtie cd stringtie make release make test
### Quality control and read trimming
---
- Initial quality check of raw reads
- Quality-trimming of reads
- Post-trimming quality check of reads
#### Initial quality check of raw reads
*FastQC is a bioinformatic tool that generates sequence quality information of your reads. Multiqc summarizes FastQC analysis logs and summarizes results in an html report.*
# 3) Run FastQC
a) Make folders for raw FastQC results and scripts
b) Write script for checking quality with FastQC and submit as job on bluewaves
- [FastQC](http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) is a tool to help identify any discrepancies or problems in your data.
nano /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/scripts/fastqc_raw.sh
#!/bin/bash #SBATCH -t 24:00:00 #SBATCH –nodes=1 –ntasks-per-node=1 #SBATCH –export=NONE #SBATCH –mem=100GB #SBATCH –mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails #SBATCH –mail-user=danielle_becker@uri.edu #your email to send notifications #SBATCH –account=putnamlab #SBATCH -D /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/raw #SBATCH –error=”script_error” #if your job fails, the error report will be put in this file #SBATCH –output=”output_script” #once your job is completed, any final job report comments will be put in this file
module load FastQC/0.11.8-Java-1.8
for file in /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/raw/*.gz do fastqc $file –outdir /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/raw/qc done
sbatch /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/scripts/fastqc_raw.sh
Submitted batch job 1816766 on 20210104
c) Make sure all files were processed
ls -1 | wc -l #64
#### Combine QC output into 1 file with MultiQC
- [MultiQC](https://multiqc.info) works with FastQC output to aggregate and present results in user-friendly way.
module load MultiQC/1.7-foss-2018b-Python-2.7.15 multiqc /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/raw/qc
c) Copy MultiQC files from the local host securely copy the MultiQC report to a local directory. Because the remote server doesn't recognize my local file paths, I need to open another terminal window and do it from there.
scp -r danielle_becker@bluewaves.uri.edu:/data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/raw/qc/*.html /Users/Danielle/Desktop/Putnam_Lab/Becker_E5/RNASeq/qc
# 4) Quality-trimming of reads
*To clean our reads we will be using a program called FastP, a tool designed to provide fast all-in-one preprocessing for FastQ files.*
++Goals of quality trimming++:
- Remove adapters
- Remove low-quality reads
- Remove reads with high abundance of unknown bases
a) Make trimmed reads folder in all other results folders
mkdir data/trimmed cd trimmed
c) Write script for trimming and run on bluewaves
++FastP Arguments/Options Used++:
- --in1 - Path to forward read input
- --in2 - Path to reverse read input
- --out1 - Path to forward read output
- --out2 - Path to reservse read output
- --failed_out - Specify file to store reads that fail filters
- --qualified_quality_phred - Phred quality >= -q is qualified (20)
- --unqualified_percent_limit - % of bases allowed to be unqualified (10)
- --length_required - Set required sequence length (100)
- --detect_adapter_for_pe - Adapters can be trimmed by overlap analysis, however, --detect_adapter_for_pe will usually result in slightly cleaner output than overlap detection alone. This results in a slightly slower run time
- --cut_right - Move a sliding window from front to tail. Use cut_right_window_size to set the window size (5), and cut_right_mean_quality (20) to set the mean quality threshold.
- --html - The html format report file name
nano /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/scripts/trim.sh
#!/bin/bash #SBATCH -t 24:00:00 #SBATCH –nodes=1 –ntasks-per-node=1 #SBATCH –export=NONE #SBATCH –mem=100GB #SBATCH –mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails #SBATCH –mail-user=danielle_becker@uri.edu #your email to send notifications #SBATCH –account=putnamlab #SBATCH -D /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/raw #SBATCH –error=”script_error” #if your job fails, the error report will be put in this file #SBATCH –output=”output_script” #once your job is completed, any final job report comments will be put in this file
module load fastp/0.19.7-foss-2018b
array1=($(ls *.fastq.gz)) #Make an array of sequences to trim for i in ${array1[@]}; do #Make a loop that trims each file in the array fastp –in1 ${i} –in2 $(echo ${i}|sed s/_R1/_R2/) –out1 ../trimmed/${i} –out2 ../trimmed/$(echo ${i}|sed s/_R1/_R2/) –qualified_quality_phred 20 –unqualified_percent_limit 10 –length_required 100 detect_adapter_for_pe –cut_right cut_right_window_size 5 cut_right_mean_quality 20 done
sbatch /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/scripts/trim.sh Submitted batch job 1819574
# 5) Post-trimming quality check of reads
Now that we've trimmed the adapters, low-quality reads and reads with many unknown bases, we will again check our sequence quality. first, we will check the trimmed sequence lengths, and then run FastQC again to examine our GC and adapter content, and our phred quality scores.
a) Check the number of files.
ls -1 | wc -l #64
b) Check the clean read count.
- [x] ++Clean Read Counts++:
zgrep -c “@GWNJ” *.gz > trimmed_seq_counts
C21_R1_001.fastq.gz:12569436 C21_R2_001.fastq.gz:12569436 C23_R1_001.fastq.gz:15920237 C18_R1_001.fastq.gz:17448563 C23_R2_001.fastq.gz:15920237 C22_R1_001.fastq.gz:17783162 C20_R1_001.fastq.gz:18346189 C22_R2_001.fastq.gz:17783162 C26_R2_001.fastq.gz:17679573 C18_R2_001.fastq.gz:17448563 C24_R1_001.fastq.gz:17244418 C26_R1_001.fastq.gz:17679573 C19_R2_001.fastq.gz:18977336 C24_R2_001.fastq.gz:17244418 C20_R2_001.fastq.gz:18346189 C19_R1_001.fastq.gz:18977336 C17_R1_001.fastq.gz:19053464 C17_R2_001.fastq.gz:19053464 C25_R1_001.fastq.gz:22683683 C25_R2_001.fastq.gz:22683683 C29_R1_001.fastq.gz:13714128 C27_R1_001.fastq.gz:18756419 C29_R2_001.fastq.gz:13714128 C27_R2_001.fastq.gz:18756419 C28_R1_001.fastq.gz:18131021 E11_R1_001.fastq.gz:14185860 E11_R2_001.fastq.gz:14185860 C28_R2_001.fastq.gz:18131021 C31_R1_001.fastq.gz:18796208 C30_R1_001.fastq.gz:20242216 C31_R2_001.fastq.gz:18796208 C30_R2_001.fastq.gz:20242216 C32_R1_001.fastq.gz:20583523 C32_R2_001.fastq.gz:20583523 E10_R1_001.fastq.gz:22441661 E12_R1_001.fastq.gz:21608172 E10_R2_001.fastq.gz:22441661 E13_R1_001.fastq.gz:18961264 E12_R2_001.fastq.gz:21608172 E13_R2_001.fastq.gz:18961264 E14_R1_001.fastq.gz:17170729 E14_R2_001.fastq.gz:17170729 E15_R1_001.fastq.gz:17920244 E15_R2_001.fastq.gz:17920244 E1_R1_001.fastq.gz:19467393 E5_R1_001.fastq.gz:12558989 E1_R2_001.fastq.gz:19467393 E4_R1_001.fastq.gz:16639960 E16_R1_001.fastq.gz:22697805 E5_R2_001.fastq.gz:12558989 E2_R2_001.fastq.gz:18593178 E4_R2_001.fastq.gz:16639960 E3_R1_001.fastq.gz:19740214 E2_R1_001.fastq.gz:18593178 E16_R2_001.fastq.gz:22697805 E3_R2_001.fastq.gz:19740214 E7_R1_001.fastq.gz:18431790 E6_R1_001.fastq.gz:18968203 E6_R2_001.fastq.gz:18968203 E7_R2_001.fastq.gz:18431790 E8_R1_001.fastq.gz:17970740 E8_R2_001.fastq.gz:17970740 E9_R1_001.fastq.gz:19333946 E9_R2_001.fastq.gz:19333946
c) Run FastQC on trimmed data
mkdir trimmed_qc
nano /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/scripts/fastqc_trimmed.sh
#!/bin/bash #SBATCH -t 24:00:00 #SBATCH –nodes=1 –ntasks-per-node=1 #SBATCH –export=NONE #SBATCH –mem=100GB #SBATCH –mail-type=BEGIN,END,FAIL #SBATCH –mail-user=danielle_becker@uri.edu #SBATCH –account=putnamlab #SBATCH -D /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/trimmed/trimmed_qc #SBATCH –error=”script_error” #SBATCH –output=”output_script”
module load FastQC/0.11.8-Java-1.8
for file in /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/trimmed/*.gz do fastqc $file –outdir /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/trimmed/trimmed_qc done
sbatch /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/scripts/fastqc_trimmed.sh Submitted batch job 1834516
d) Run MultiQC on trimmed data
module load MultiQC/1.7-foss-2018b-Python-2.7.15 multiqc /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/trimmed/trimmed_qc
e) Copy MultiQC files from the local host securely copy the MultiQC report to a local directory. Because the remote server doesn't recognize my local file paths, I need to open another terminal window and do it from there.
scp -r danielle_becker@bluewaves.uri.edu:/data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/trimmed/trimmed_qc/*.html /Users/Danielle/Desktop/Putnam_Lab/Becker_E5/RNASeq/trimmed_qc
# 6) Alignment of clean reads to reference genome
---
*HISAT2 is a fast and sensitive alignment program for mapping next-generation DNA and RNA sequencing reads to a reference genome.*
- Index the reference genome
- Alignment of clean reads to the reference genome
a) Generate genome build
### Need to unzip genome files before running
gunzip Pver_genome_assembly_v1.0.fasta.gz gunzip Pver_genome_assembly_v1.0.gff3.gz
### HiSat2 Align reads to reference genome
b) Index the reference genome
Index the reference genome in the reference directory.
++HISAT2-build Alignment Arguments Used++:
- <reference_in> - name of reference files
- <gt2_base> - basename of index files to write
- -f - reference file is a FASTA file
nano /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/scripts/Hisat2_genome_build.sh
#!/bin/bash #SBATCH -t 24:00:00 #SBATCH –nodes=1 –ntasks-per-node=1 #SBATCH –export=NONE #SBATCH –mem=100GB #SBATCH –mail-type=BEGIN,END,FAIL #SBATCH –mail-user=danielle_becker@uri.edu #SBATCH –account=putnamlab #SBATCH -D /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/refs #SBATCH –error=”script_error” #SBATCH –output=”output_script”
module load HISAT2/2.1.0-foss-2018b
hisat2-build -f /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/refs/Pverr/Pver_genome_assembly_v1.0.fasta ./Pver_ref
sbatch /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/scripts/Hisat2_genome_build.sh Submitted batch job 1834918
c) Alignment of clean reads to the reference genome
Align your reads to the index files. We will do this by writing a script we will call ```Hisat2_align2.sh```. This script will also take the output SAM files from our HISAT2 alignment and covert them into the sorted BAM files that are the necessary input for our assembly tool, StringTie. We do this by calling SAMtools in our script.
++HISAT2 Alignment Arguments Used++:
- -x <hisat2-idx> - Basename of index files to read
- -1 <m1> - List of forward sequence files
- -2 <m1> - List of reverse sequence files
- -S - Name of output files
- -q - Input files are in FASTQ format
- -p - Number processors
- --rf - Reads are stranded
- --dta - Adds the XS tag to indicate the genomic strand that produced the RNA from which the read was sequenced. As noted by StringTie... "be sure to run HISAT2 with the --dta option for alignment, or your results will suffer."
++SAMtools Options Arguments Used++:
- -@ - Number threads
- -o - Output file
mkdir mapped
nano /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/scripts/Hisat2_align2.sh
#!/bin/bash #SBATCH -t 72:00:00 #SBATCH –nodes=1 –ntasks-per-node=5 #SBATCH –export=NONE #SBATCH –mem=500GB #SBATCH –mail-type=BEGIN,END,FAIL #SBATCH –mail-user=danielle_becker@uri.edu #SBATCH –account=putnamlab #SBATCH -D /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/trimmed #SBATCH -p putnamlab #SBATCH –cpus-per-task=3 #SBATCH –error=”script_error” #SBATCH –output=”output_script”
module load HISAT2/2.1.0-foss-2018b
#Aligning paired end reads #Has the R1 in array1 because the sed in the for loop changes it to an R2. SAM files are of both forward and reverse reads
array1=($(ls *_R1_001.fastq.gz))
for i in ${array1[@]}; do
hisat2 -p 48 –rna-strandness RF –dta -q -x /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/refs/Pver_ref -1 /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/trimmed/${i}
-2 /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/trimmed/$(echo ${i}|sed s/_R1/_R2/) -S /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/mapped/${i}.sam
done
sbatch /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/scripts/Hisat2_align2.sh #Submitted batch job 19930
## Sort and convert sam to bam
nano /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/scripts/SAMtoBAM.sh
#There will be lots of .tmp file versions in your folder, this is normal while this script runs and they should delete at the end to make one sorted.bam file
#!/bin/bash #SBATCH -t 72:00:00 #SBATCH –nodes=1 –ntasks-per-node=8 #SBATCH –export=NONE #SBATCH –mem=500GB #SBATCH –mail-type=BEGIN,END,FAIL #SBATCH –mail-user=danielle_becker@uri.edu #SBATCH –account=putnamlab #SBATCH -D /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/mapped #SBATCH –cpus-per-task=3 #SBATCH –error=”script_error” #SBATCH –output=”output_script”
module load SAMtools/1.9-foss-2018b
array1=($(ls *.sam)) for i in ${array1[@]}; do samtools sort -o /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/mapped/${i}.sorted.bam /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/mapped/${i} done
sbatch /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/scripts/SAMtoBAM.sh #Submitted batch job 19931
### Remove Sam files to save space
rm /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/mapped/*.sam
### Check number of mapped reads
zgrep -c “@GWNJ” *.bam > mapped_reads_counts
Now we've got some sorted BAM files that can be used in our assembly!!
# 7) Assemble aligned reads and quantify transcripts
---
*StringTie is a fast and highly efficient assembler of RNA-Seq alignments into potential transcripts.*
- Reference-guided assembly with novel transcript discovery
- Merge output GTF files and assess the assembly performance
- Compilation of GTF-files into gene and transcript count matrices
### Needed to modify Pverr_genome_assembly file, information in this [GitHub issue](https://github.com/Putnam-Lab/Lab_Management/issues/11)
##copy modified genome assembly file to bluewaves, enter this command into local computer shell
scp -r /Users/Danielle/Downloads/Pver_genome_assembly_v1.0_modified.gff3 danielle_becker@bluewaves.uri.edu:/data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/refs/Pverr/
mkdir counts cd counts
Create the StringTie reference-guided assembly script, ```StringTie_Assemble.sh``` *
++StringTie Arguments Used++:
- -A - Output gene abundance file
- -p - Specify number of processers
- --rf - Reads are stranded
- -e - Limit the estimation and output of transcripts to only those that match the reference (in this case, our merged GTF)
- -G - Specify annotation file
- -o - Name of output file
b) Assemble and estimate reads
nano /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/scripts/StringTie_Assemble.sh
#!/bin/bash #SBATCH -t 72:00:00 #SBATCH –nodes=1 –ntasks-per-node=5 #SBATCH –export=NONE #SBATCH –mail-type=BEGIN,END,FAIL #SBATCH –mail-user=danielle_becker@uri.edu #SBATCH –account=putnamlab #SBATCH -D /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/mapped #SBATCH –cpus-per-task=3
module load StringTie/2.1.4-GCC-9.3.0
array1=($(ls *.bam)) for i in ${array1[@]}; do stringtie -p 48 –rf -e -G /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/refs/Pverr/Pver_genome_assembly_v1.0_modified.gff3 -o /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/counts/${i}.gtf /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/mapped/${i} done
sbatch /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/scripts/StringTie_Assemble.sh Submitted batch job 1871636
d) Merge stringTie gtf results
- *Gffcompare is a tool that can compare, merge, annotate and estimate accuracy of GFF/GTF files when compared with a reference annotation*
- In this step we are making a file with all the gtf names and stringtie will merge them all together for a master list for your specific genes
- Using the StringTie merge mode, merge the assembly-generated GTF files to assess how well the predicted transcripts track to the reference annotation file. This step requires the TXT file, ```mergelist.txt```. This file lists all of the file names to be merged. *Make sure ```mergelist.txt``` is in the StringTie program directory*.
++StringTie Arguments Used++:
- --merge - Distinct from the assembly usage mode used above, in the merge mode, StringTie takes as input a list of GTF/GFF files and merges/assembles these transcripts into a non-redundant set of transcripts.
- -p - Specify number of processers
- -G - Specify reference annotation file. With this option, StringTie assembles the transfrags from the input GTF files with the reference sequences
- -o - Name of output file
- <mergelist.txt> - File listing all filenames to be merged. Include full path.
ls *gtf > mergelist.txt cat mergelist.txt
module load StringTie/2.1.4-GCC-9.3.0
stringtie –merge -p 8 -G /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/refs/Pverr/Pver_genome_assembly_v1.0_modified.gff3 -o stringtie_merged.gtf mergelist.txt
e) Assess the performance of the assembly
Now we can use the program gffcompare to compare the merged GTF to our reference genome.
++Gffcompare Arguments Used++:
- -r - Specify reference annotation file
- -G - Compare all the transcripts in our input file ```stringtie_merged.gtf```
- -o - Prefix of all output files
module load gffcompare/0.11.5-foss-2018b
gffcompare -r /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/refs/Pverr/Pver_genome_assembly_v1.0_modified.gff3 -o merged stringtie_merged.gtf
Some of the output files you will see are...
- merged.stats
- merged.tracking
- merged.annotated.gtf
- merged.stringtie_merged.gtf.refmap
- merged.loci
- merged.stringtie_merged.gtf.tmap
We are most interested in the files ```merged.annotation.gtf``` and ```merged.stats```. The file ```merged.annotation.gtf``` tells you how well the predicted transcripts track to the reference annotation file and the file ```merged.stats``` file shows the sensitivity and precision statistics and total number for different features (genes, exons, transcripts). Then, from the local host securely copy ```merged.stats``` to a local directory. Unfortunately, ```merged.annotation.gtf``` is too big to store locally, but we can view it remotely.
e) Re-estimate assembly
nano re_estimate.assembly.sh
#!/bin/bash #SBATCH -t 72:00:00 #SBATCH –nodes=1 –ntasks-per-node=5 #SBATCH –export=NONE #SBATCH –mail-type=BEGIN,END,FAIL #SBATCH –mail-user=danielle_becker@uri.edu #SBATCH –account=putnamlab #SBATCH -D /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/mapped #SBATCH –cpus-per-task=3
module load StringTie/2.1.4-GCC-9.3.0
array1=($(ls *.bam)) for i in ${array1[@]}; do stringtie -e -G /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/refs/Pverr/Pver_genome_assembly_v1.0_modified.gff3 -o ${i}.merge.gtf ${i} echo “${i}” done
sbatch /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/scripts/re_estimate.assembly.sh Submitted batch job 1871641
move merged GTF files to their own folder
mv *merge.gtf ../GTF_merge
f) Compilation of GTF-files into gene and transcript count matrices
The StringTie program includes a script, ```prepDE.py``` that compiles your assembly files into gene and transcript count matrices. This script requires as input the list of sample names and their full file paths, [```sample_list.txt```](https://github.com/echille/Montipora_OA_Development_Timeseries/blob/master/RNAseq_Analyses/sample_list.txt). This file will live in StringTie program directory.
Go back into your stringtie directory (the one I should have named assembly). Run ```prepDE.py``` to merge assembled files together into a DESeq2-friendly version.
++StringTie prepDE.py Arguments Used++:
- -i - Specify that input is a TXT file
- -g - Require output gene count file, default name is ```gene_count_matrix.csv```
- -t - Require output transcript count gene count file, default name is ```transcript_count_matrix.csv```
#making a sample txt file with all gtf file names
F=/data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/mapped/GTF_merge/
array2=($(ls *merge.gtf)) for i in ${array2[@]} do echo “${i} $F${i}” » sample_list.txt done
#sample_list.txt document
C17_R1_001.fastq.gz.sam.sorted.bam.merge.gtf /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/mapped/GTF_merge/C17_R1_001.fastq.gz.sam.sorted.bam.merge.gtf C18_R1_001.fastq.gz.sam.sorted.bam.merge.gtf /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/mapped/GTF_merge/C18_R1_001.fastq.gz.sam.sorted.bam.merge.gtf C19_R1_001.fastq.gz.sam.sorted.bam.merge.gtf /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/mapped/GTF_merge/C19_R1_001.fastq.gz.sam.sorted.bam.merge.gtf C20_R1_001.fastq.gz.sam.sorted.bam.merge.gtf /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/mapped/GTF_merge/C20_R1_001.fastq.gz.sam.sorted.bam.merge.gtf C21_R1_001.fastq.gz.sam.sorted.bam.merge.gtf /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/mapped/GTF_merge/C21_R1_001.fastq.gz.sam.sorted.bam.merge.gtf C22_R1_001.fastq.gz.sam.sorted.bam.merge.gtf /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/mapped/GTF_merge/C22_R1_001.fastq.gz.sam.sorted.bam.merge.gtf C23_R1_001.fastq.gz.sam.sorted.bam.merge.gtf /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/mapped/GTF_merge/C23_R1_001.fastq.gz.sam.sorted.bam.merge.gtf C24_R1_001.fastq.gz.sam.sorted.bam.merge.gtf /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/mapped/GTF_merge/C24_R1_001.fastq.gz.sam.sorted.bam.merge.gtf C25_R1_001.fastq.gz.sam.sorted.bam.merge.gtf /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/mapped/GTF_merge/C25_R1_001.fastq.gz.sam.sorted.bam.merge.gtf C26_R1_001.fastq.gz.sam.sorted.bam.merge.gtf /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/mapped/GTF_merge/C26_R1_001.fastq.gz.sam.sorted.bam.merge.gtf C27_R1_001.fastq.gz.sam.sorted.bam.merge.gtf /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/mapped/GTF_merge/C27_R1_001.fastq.gz.sam.sorted.bam.merge.gtf C28_R1_001.fastq.gz.sam.sorted.bam.merge.gtf /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/mapped/GTF_merge/C28_R1_001.fastq.gz.sam.sorted.bam.merge.gtf C29_R1_001.fastq.gz.sam.sorted.bam.merge.gtf /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/mapped/GTF_merge/C29_R1_001.fastq.gz.sam.sorted.bam.merge.gtf C30_R1_001.fastq.gz.sam.sorted.bam.merge.gtf /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/mapped/GTF_merge/C30_R1_001.fastq.gz.sam.sorted.bam.merge.gtf C31_R1_001.fastq.gz.sam.sorted.bam.merge.gtf /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/mapped/GTF_merge/C31_R1_001.fastq.gz.sam.sorted.bam.merge.gtf C32_R1_001.fastq.gz.sam.sorted.bam.merge.gtf /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/mapped/GTF_merge/C32_R1_001.fastq.gz.sam.sorted.bam.merge.gtf E10_R1_001.fastq.gz.sam.sorted.bam.merge.gtf /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/mapped/GTF_merge/E10_R1_001.fastq.gz.sam.sorted.bam.merge.gtf E11_R1_001.fastq.gz.sam.sorted.bam.merge.gtf /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/mapped/GTF_merge/E11_R1_001.fastq.gz.sam.sorted.bam.merge.gtf E12_R1_001.fastq.gz.sam.sorted.bam.merge.gtf /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/mapped/GTF_merge/E12_R1_001.fastq.gz.sam.sorted.bam.merge.gtf E13_R1_001.fastq.gz.sam.sorted.bam.merge.gtf /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/mapped/GTF_merge/E13_R1_001.fastq.gz.sam.sorted.bam.merge.gtf E14_R1_001.fastq.gz.sam.sorted.bam.merge.gtf /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/mapped/GTF_merge/E14_R1_001.fastq.gz.sam.sorted.bam.merge.gtf E15_R1_001.fastq.gz.sam.sorted.bam.merge.gtf /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/mapped/GTF_merge/E15_R1_001.fastq.gz.sam.sorted.bam.merge.gtf E16_R1_001.fastq.gz.sam.sorted.bam.merge.gtf /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/mapped/GTF_merge/E16_R1_001.fastq.gz.sam.sorted.bam.merge.gtf E1_R1_001.fastq.gz.sam.sorted.bam.merge.gtf /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/mapped/GTF_merge/E1_R1_001.fastq.gz.sam.sorted.bam.merge.gtf E2_R1_001.fastq.gz.sam.sorted.bam.merge.gtf /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/mapped/GTF_merge/E2_R1_001.fastq.gz.sam.sorted.bam.merge.gtf E3_R1_001.fastq.gz.sam.sorted.bam.merge.gtf /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/mapped/GTF_merge/E3_R1_001.fastq.gz.sam.sorted.bam.merge.gtf E4_R1_001.fastq.gz.sam.sorted.bam.merge.gtf /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/mapped/GTF_merge/E4_R1_001.fastq.gz.sam.sorted.bam.merge.gtf E5_R1_001.fastq.gz.sam.sorted.bam.merge.gtf /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/mapped/GTF_merge/E5_R1_001.fastq.gz.sam.sorted.bam.merge.gtf E6_R1_001.fastq.gz.sam.sorted.bam.merge.gtf /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/mapped/GTF_merge/E6_R1_001.fastq.gz.sam.sorted.bam.merge.gtf E7_R1_001.fastq.gz.sam.sorted.bam.merge.gtf /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/mapped/GTF_merge/E7_R1_001.fastq.gz.sam.sorted.bam.merge.gtf E8_R1_001.fastq.gz.sam.sorted.bam.merge.gtf /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/mapped/GTF_merge/E8_R1_001.fastq.gz.sam.sorted.bam.merge.gtf E9_R1_001.fastq.gz.sam.sorted.bam.merge.gtf /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/mapped/GTF_merge/E9_R1_001.fastq.gz.sam.sorted.bam.merge.gtf
#create gene matrix
nano /data/putnamlab/hputnam/Becker_E5/RNASeq_Becker_E5/scripts/GTFtoCounts.sh
#!/bin/bash #SBATCH -t 72:00:00 #SBATCH –nodes=1 –ntasks-per-node=5 #SBATCH –export=NONE #SBATCH –mail-type=BEGIN,END,FAIL #SBATCH –mail-user=danielle_becker@uri.edu #SBATCH –account=putnamlab #SBATCH -D /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/mapped/GTF_merge #SBATCH –cpus-per-task=3
module load StringTie/2.1.4-GCC-9.3.0 module load Python/2.7.18-GCCcore-9.3.0
python prepDE.py -g Poc_gene_count_matrix.csv -i sample_list.txt
sbatch /data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/scripts/GTFtoCounts.sh Submitted batch job 1871645
g) Finally, move your count matrices into the output directory and securely copy them to your local directory, from your local host.
#copy gene count matrix
scp danielle_becker@bluewaves.uri.edu:/data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/mapped/GTF_merge/Poc_gene_count_matrix.csv /Users/Danielle/Desktop/Putnam_Lab/Becker_E5/RNASeq/
#copy transcript count matrix scp danielle_becker@bluewaves.uri.edu:/data/putnamlab/dbecks/Becker_E5/Becker_RNASeq/data/mapped/GTF_merge/transcript_count_matrix.csv /Users/Danielle/Desktop/Putnam_Lab/Becker_E5/RNASeq/
```